The only thing that change between lesion masks and the disconnectome maps (format-wise I mean) is the values in the voxels that are continuous instead of binary. So, if you binarised your disconnectome maps they should work with AnaCOM2.
Otherwise, you can send me some of your disconnectome maps so I can have a look. Also, the log file would be useful or any error message you got.
I have a problem getting the AnaCOM2 to create a full set of output files;
I have a set of nifty files containing lesion segmentations in MNI152 space. With naming; subject_2017008.nii, subject_2017010.nii and so on. I have a .csv file containing two columns, one with the names and one with results from a neuropsychological test. The file is without headers.
I have chosen the following: Mann Whitney test, overlap threshold 3, published normative value and keep temporary files.
There are no error or warning messages.
In the output file I then get; copypatients.csv, logAnacom.txt, patients_info.csv and a directory called anacomTemporaryFiles containing; maskedStd.nii.gz, maskedMeanValMap.nii.gz and a directory called anacomClustersDir.
However, I expected several more files, containing results from statistical tests, like clusters.csv, clusters.nii.gz and clusters_holm.nii.gz.
The problem might be the fact that the patients_info.csv file contains NULL for the cells where I would expect names of the subjects. I have tried different naming conventions but always get same results. I have tried looking in the logAnacom.txt file but can’t find anything about the patients_info.csv
Version:
I am running the BCBtoolkit for Mac on a Mac Ventura 13.2.1.
Relevant log outputs:
The last part of the log output is:
+(410)ph_csv=/Users/berghelland/Documents/Gisle/Forskning/Egne_artikler/Ph.D._Artikler/Cognition_model/MRI/Executive/Anacom/output/clusters.csv
+(412)‘[’ 1 ‘!=’ 1 ‘]’
+(480)[[ -e /Users/berghelland/Documents/Gisle/Forskning/Egne_artikler/Ph.D._Artikler/Cognition_model/MRI/Executive/Anacom/output/anacomTemporaryFiles ]]
+(482)mv /Users/berghelland/Documents/Gisle/Forskning/Egne_artikler/Ph.D._Artikler/Cognition_model/MRI/Executive/Anacom/output/anacomClustersDir /Users/berghelland/Documents/Gisle/Forskning/Egne_artikler/Ph.D._Artikler/Cognition_model/MRI/Executive/Anacom/output/anacomTemporaryFiles
+(483)mv /Users/berghelland/Desktop/Converstions_to_MNI/BCBToolKitOSX/Tools/tmp/tmpAnacom/maskedStd.nii.gz /Users/berghelland/Documents/Gisle/Forskning/Egne_artikler/Ph.D._Artikler/Cognition_model/MRI/Executive/Anacom/output/anacomTemporaryFiles
+(484)mv /Users/berghelland/Desktop/Converstions_to_MNI/BCBToolKitOSX/Tools/tmp/tmpAnacom/maskedMeanValMap.nii.gz /Users/berghelland/Documents/Gisle/Forskning/Egne_artikler/Ph.D._Artikler/Cognition_model/MRI/Executive/Anacom/output/anacomTemporaryFiles
+(488)echo ‘#’
Hope you have suggestions for a solution or further troubleshooting?
We have been having some issues with the latest OS versions and we tried some modifications to the scripts. You can try to replace the following scripts in your version of the BCBToolkit:
It has worked for some people but not everyone. When I have a bit of time I will make a proper update to AnaCOM2 to make it faster and more reliable on different systems.
In the mean time, I hope that helps. Otherwise, you can have a look at FSL randomise tools which also has a cluster-based analysis.
I tried with the modified scripts. Unfortunately it did not work. I then tried running on Ubuntu. The process got a bit further - but would still not make the clusters.csv file. I modified the script and it is now making the file, but it is either not doing the analysis properly or not writing the file properly, since output of the statistical tests (pval, stat and holm) is NA. I will continue to troubleshoot the script, and run FSL randomise simultaneously - thanks for the tip.
I am trying to run the disconnectome analysis on BCBToolkit and then motor prediction using the Disconnectome Symptom Discoverer.
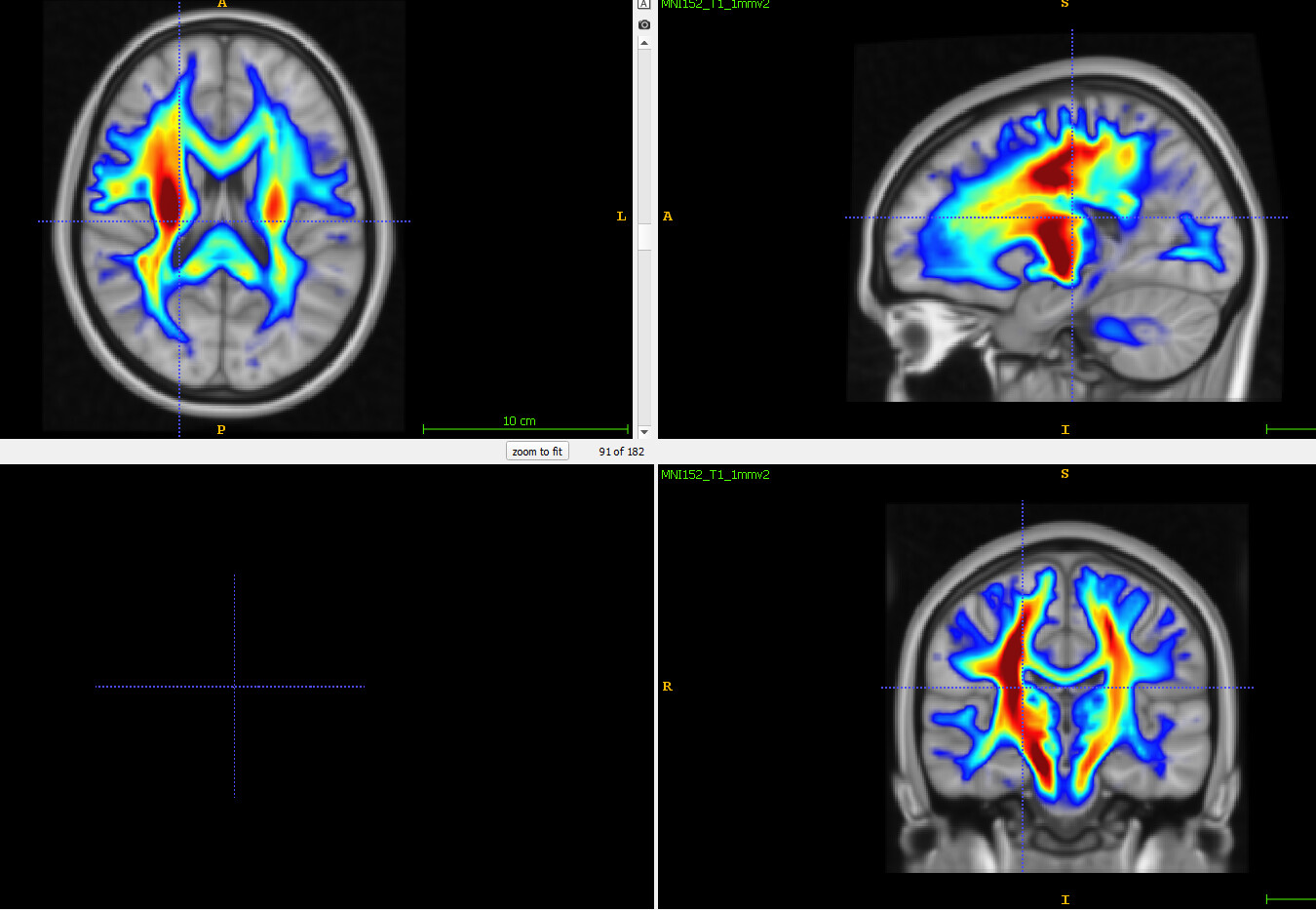
The disconnectome analysis on BCBToolkit works fine when using lesions in 1mm MNI space, outputting disconnectome also in 1mm MNI space, however when I input the same lesions resampled to 2mm MNI space, the output disconnectomes are all zeros. The 2mm and 1mm lesions overlap when overlayed, and the 2mm lesions have the same image dimensions as the sample disconnectome provided so the registration should be fine.
For the Disconnectome Symptom Discoverer, I used a workaround by resampling the 1mm disconnectomes to 2mm, then using as input. However, some motor score predictions (particularly ARAT) are almost all the same across individuals (e.g. both L and R ARAT grasp ranges between 16-18 in basically all participants) even though the UMAP coordinates differ. I was wondering if this might be due to the resampling process, rather than starting from 2mm lesions, but the disconnectomes do look sensible and fit correctly when overlayed against MNI template. Is this expected behaviour or have I done something wrong?
To run the disconnectome exactly as in Talozzi et al., please visit our website at http://www.bcblab.com/ and download the DISCONNECTOME Package X (which includes 180 additional subjects from HCP7T at 2mm resolution) from the Open Data section. Replace the .trk files in the BCBtoolkit directory (located at BCBtoolkit/Tools/extrafiles/tracks) with the ones you just downloaded.
You can then use 2mm lesions directly in BCBtoolkit.
Regarding the DSD predictions, if your patient does not fall into a risk territory on the UMAP associated with a poor outcome at 1 year, the DSD will predict the maximum score. This prediction remains consistent even if patients have different coordinates within the UMAP. Does that make sense?
I think I fixed the 2mm analysis as instructed but have not completed it for my dataset yet as it takes much longer, I’m guessing due to the large number of .trk files.
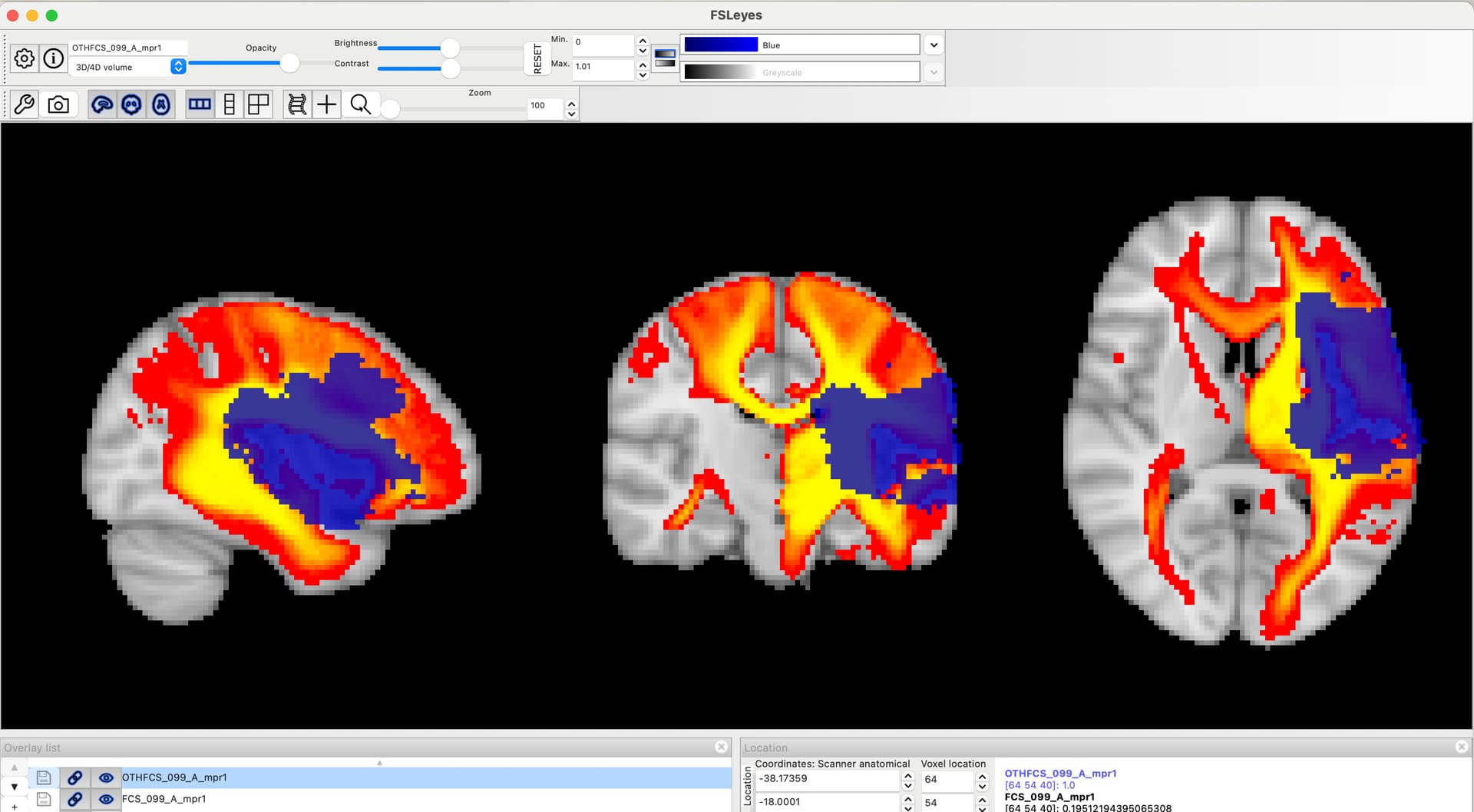
For the DSD predictions, I understand, but the predictions for our dataset still feel consistently high even though our dataset has plenty of variation in lesions/disconnectomes. See screenshot of an overlay of our dataset’s disconnectomes against MNI template. Looking at the DSD article and supplementary info, I’m thinking this may be because the original dataset used to develop the DSD had mostly good ARAT outcomes?
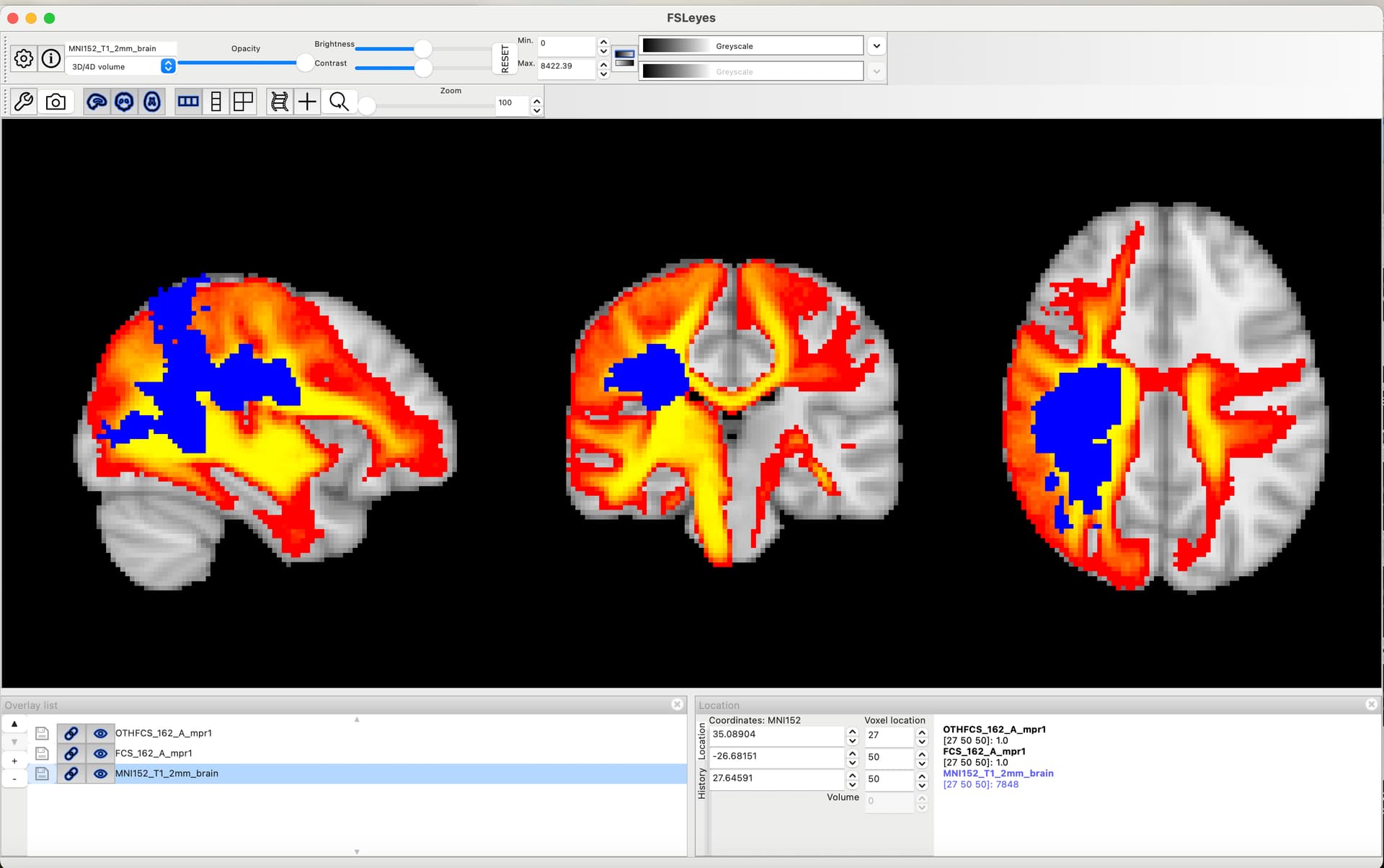
Hi Ben, regarding the ARAT (laragrasp,laragrip,larapinch,raragrasp,raragrip,rarapinch) predictions. As you can see in Supplementary Fig. 10 and 13, we have a few patients with low ARAT scores. However, their lesions are pretty big and do not cluster specifically in the disconnectome embedding space. This impacted the DSD prediction in a manner that patients presented; on average, 0 scores on the ARAT were predicted with low scores (e.g. 5, or < 10) but not 0 as in the real measure. This is probably due to the lack of a clear cluster in the morphospace and the smoothing parameter used on the patient’s morphospace coordinates. I am attaching two patients: p.99 ARAT measured (laragrasp=18,laragrip=12,larapinch=18,raragrasp=0,raragrip=0,rarapinch=0), p.099 ARAT predicted (laragrasp=17.8,laragrip=12,larapinch=17.6,raragrasp=8.47,raragrip=5.9,rarapinch=9.5). p. 162 ARAT measured (laragrasp=0,laragrip=0,larapinch=0,raragrasp=18,raragrip=12,rarapinch=18), p. 162 ARAT predicted ( laragrasp=9.4,laragrip=6.97,larapinch=7.89,raragrasp=17.7,raragrip=11.8,rarapinch=17.43). Thus, for the ARAT scale, it may be helpful to interpret the DSD prediction just as impaired or not, using a scale cut-off, e.g. predictions below 10. I hope this will be helpful. Thanks. Lia
Hi everyone! Sorry for the question but I am a beginer with Mac! I have downloaded BCB Toolkit-master but I can’t make it run. It says I should double click on the script BCBToolKit.command, but there is no such script. I only find the BCBToolKit.sh, which actually for Linux is.
I have also downloaded the BCB ToolkitOSX.zip but I can’t open the folder; the Format is not being supported.
Any ideas what I should do?
If I understand correctly you downloaded the code from github? Unfortunately, this repository is only for the source code of the BCBToolkit so, it does not contain all that is needed to run the tool.
Do you have an error message when you try to open the folder after extracting the zip file BCBToolkitOSX.zip?
Thank you very much for the reply!
yes that’s right, I have downloaded it from the GitHub.
I can not extract the zip file from BCB TooliktOSX.zip, it shows an error that the format is not supported. I cannot open either with an Unarchiver.
Hiya, I’m having similar problems other people have commented with trying to run the disconnectome module and ending up with an empty logs folder.
I’m running it with MacOS Sonoma 14.5 and downloaded it from the BCBtoolkit website. I have the most recent version of JAVA installed. I’m having the problem with the sample lesions file provided so its not just my file.
I’ve tried the following things (all together):
running the sudo spctl --master-disable
redownloading the toolkit after installing java
replacing the nscores lines with ncores=$(getconf _NPROCESSORS_ONLN), and I’ve tried leaving the original two lines and adding that one after
I’ve checked the permissions on the info on the bcbtoolkit folder and it says i have permission to read and write - is that enough?
Sorry, yes, it seems that this problem is becoming more frequent with the new versions of the operating systems. Unfortunately, I haven’t had time to fix the interface to work around this problem but I plan on updating the BCBtoolkit in the near future to fix all these issues.
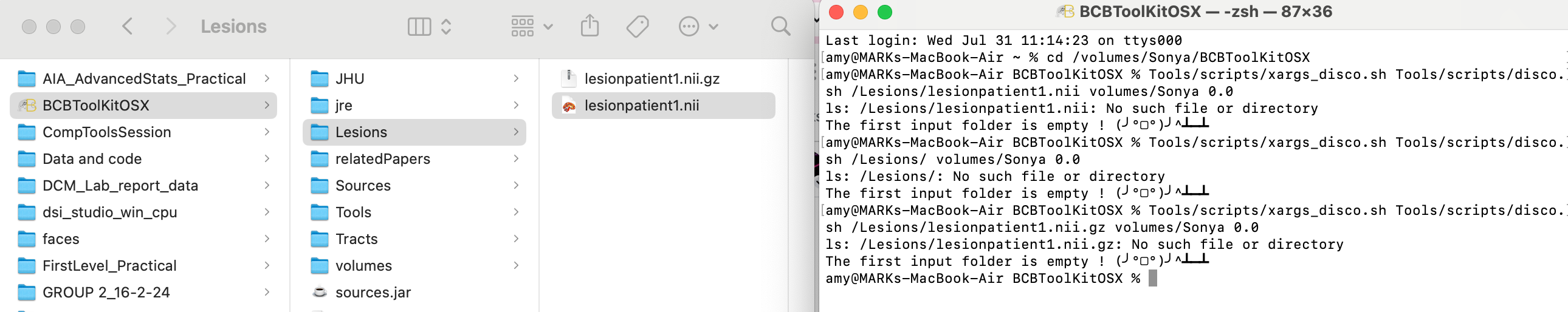
In the meantime, you can use the scripts in the terminal: xargs_disco.sh is located in BCBToolkit/Tools/scripts
cd in the BCBToolkit folder and then run:
Tools/scripts/xargs_disco.sh Tools/scripts/disco.sh path_to_inputpath_to_output 0.0
0.0 being the threshold you want (or not) to apply to the output disconnectomes.
Indeed, there seem to be a problem with the OSX archive stored on our website, we will see what’s going on and fix it as soon as we can! Sorry about that
Thank you for that! I’ve tried that and it’s now saying that the the input folder is empty - I’ve attached a screenshot of my folders and the code i’ve tried. Is there something else I’m missing?
Let me jump in here. Chris and I just tested on my M1 laptop upgraded to Sonoma today and BCBtoolkit works. Therefore the issue might be coming from M3 chip. I’ll buy an M3 in september but meanwhile, we are really sorry and we will be happy to run BCBtoolkit analyses for you. Just send us your lesions (michel.thiebaut@gmail.com) and we will process it swiftly.