I’ve been running into some unfamiliar issues with the previous two versions of fmriprep (the current one 20.2.0 and 20.2.1) where for some subjects their json files have “null” values in their confound estimates (see attached). With each of these versions it happens to a different set of subjects, making it a little challenging to troubleshoot. I did check these subjects anat folders and they have other tissue segmentations (e.g., can visualized wm and csf segmentations), so I don’t think it’s a freesurfer issue. This didn’t happen with version 1.5.1, but I know there were other bugs with that one, so I wanted to use the most up to date version. I’m thinking because of our longitudinal BIDS structure, the parent anat folder may have files overwritten since the anatomical segmentations do not land in the anat folder of the session folders. The reason I’m thinking this is bc it seems to be only a ses-T1 issue (if it’s running longitudinally through anat processing first, then the files that land in the parent anat folder come from later session segmentations?). For example,
sub-XXX
anat (all the segmentation niftis and txt files, but “ses-T” is not in any file names, which makes me think that it’s overwritten?)
log
figures
ses-T1
anat (just the orig_to-T1w_mode-image_xfm.txt)
func
ses-T1x
anat (just the orig_to-T1w_mode-image_xfm.txt)
func
ses-T2
anat (just the orig_to-T1w_mode-image_xfm.txt)
func
I’m testing out just one subject who has this structure and on only time one to see if the issue persists, but wanted to ask if anyone has run into this at any point or may have some troubleshooting advice.
Generally session information is removed from anatomical derivatives, since multiple T1w images are combined to create a common subject template. The transforms that map between the input files and the template are what you find in ses-*/anat/.
This seems likely to be a result of failure to converge showing up in the metadata rather than a crash. The most likely cause is a bad aCompCor mask, which you should be able to see in your reports. @rastko might be able to comment here…
Thank you, that helps. I’m using fmriprep version 20.2.1; however, most subjects who have this problem ran successfully under an older fmriprep version (1.5.1). I did archive their outputs from previous fmrirpep versions. So, I’m wondering if I can use the regressor/confound files from the previous version on the preproc bold data of the new version given (if I understand correctly) the confounds are generated based on the raw data.
I would not reuse confounds that were not generated by the same process that generated the final BOLD series. Although they are very likely to be correlated, I would be skeptical as a reviewer for why you trust the two pieces to work together without extensive verification that would probably be more work than trying to fix fMRIPrep to ensure it works on your data. If you’re able to share the data, we can try to reproduce the issue.
Thank you! I was able to figure most of issue out. It seems like with our longitudinal data structure, the FreeSurfer files were re-writing over the previous wave of data. We had previously run FreeSurfer on all the data, and stored those outputs elsewhere, so I was able to use those and cross-sectionally run fmriprep at each wave using the correct FreeSurfer outputs. There are still 13 subjects who have null values in all rows of csf and/or white matter (and their derivatives). However, there is nothing in the .err log to probe and the .out file ends with exit code 0. Thank you for offering to reproduce the issue. I will de-identify and share.
Are you seeing this as well? I suspect this is the root of the problem. I’m re-running with --ignore fieldmaps to see if there are other problems.
@oesteban might see something here, but my familiarity with the ways fieldmaps can fail is limited. I hope it’s just something wrong with the metadata, but it may be that the fieldmap is irretrievably broken or there’s a bug in how we apply it.
@jackie-schwartz As an update, running with --ignore fieldmaps had sensible-looking confounds. I would carefully check your fieldmap files and metadata to ensure that they are of similar quality to subjects where the problem doesn’t arise.
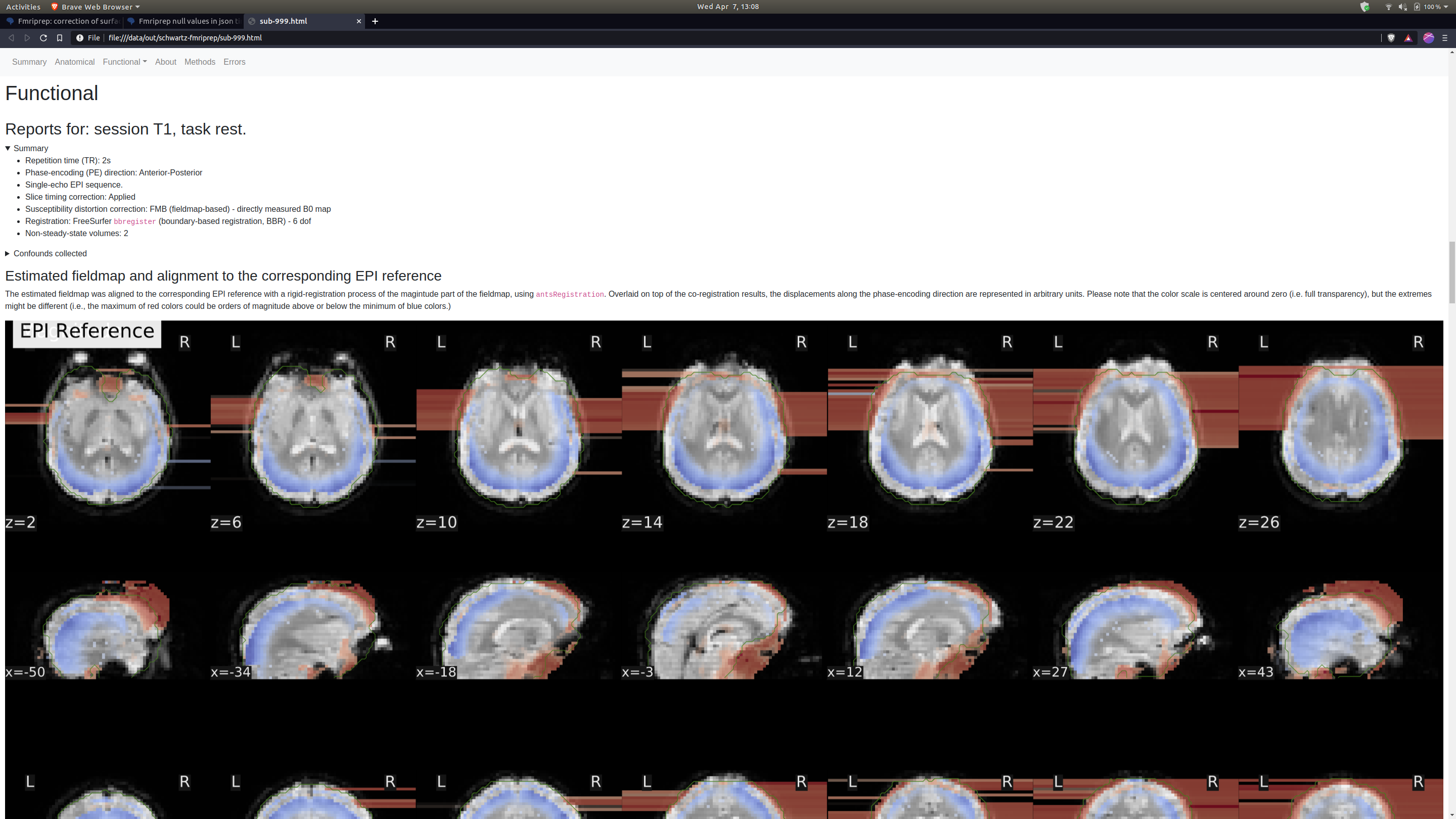
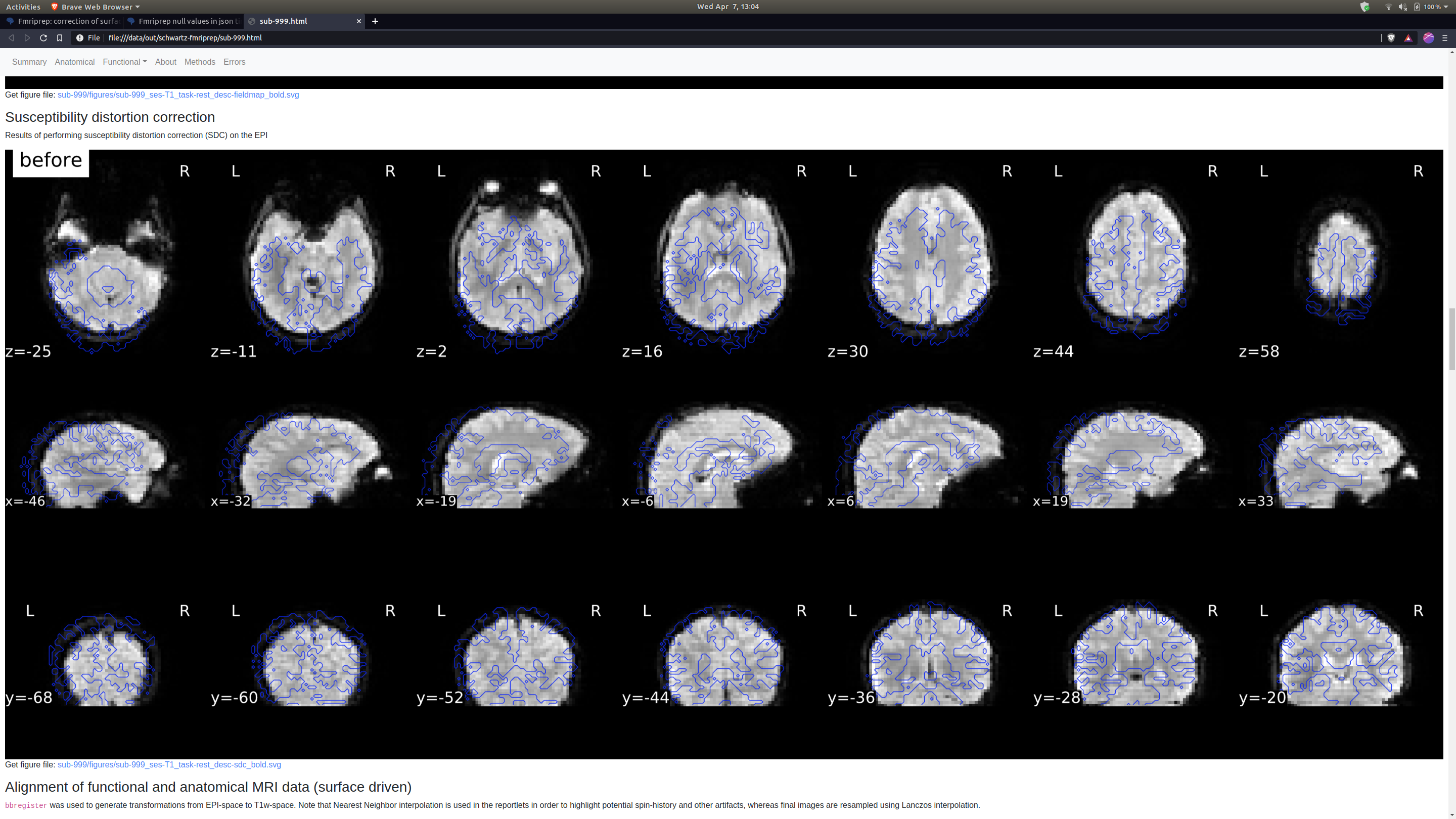
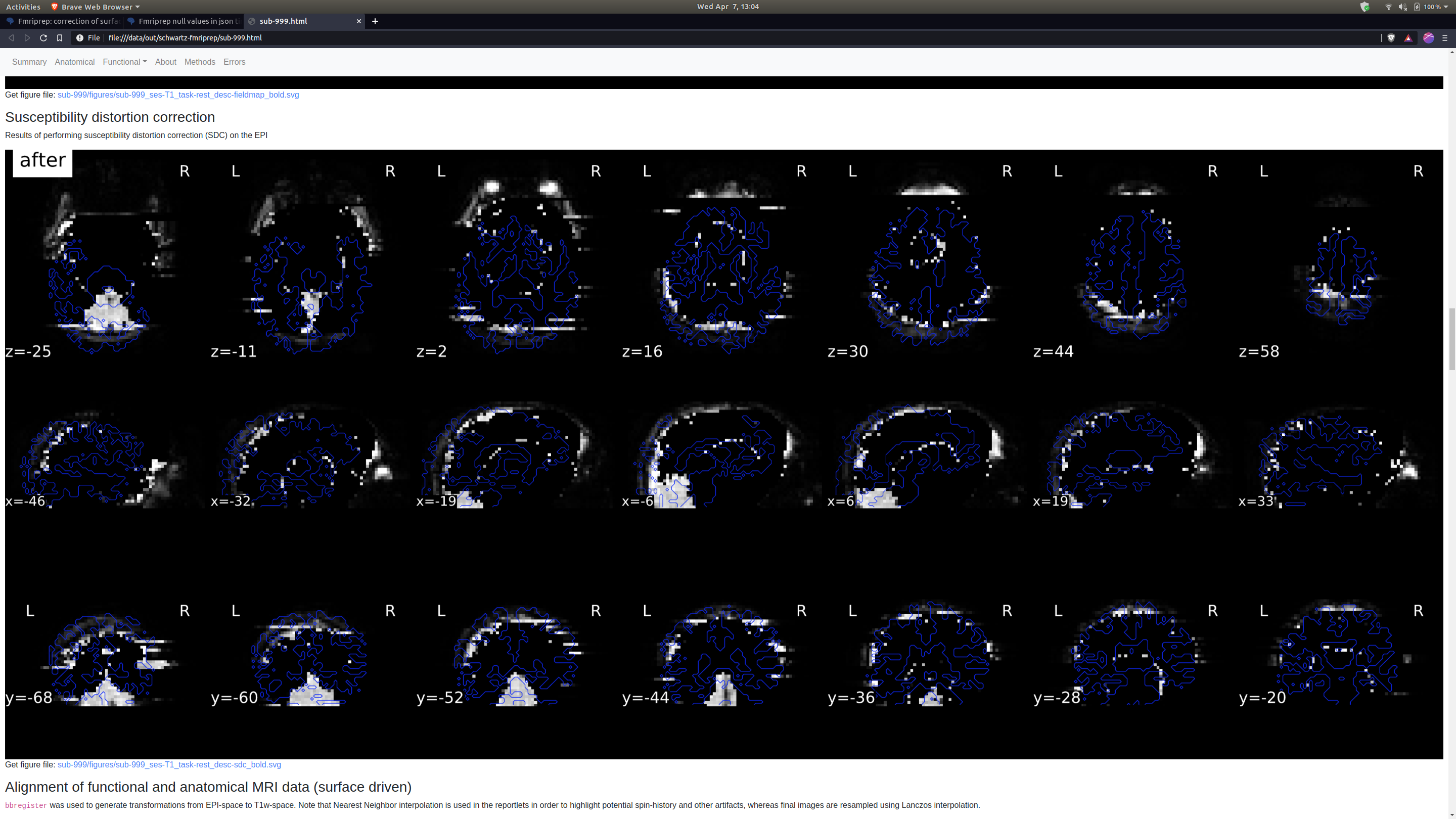
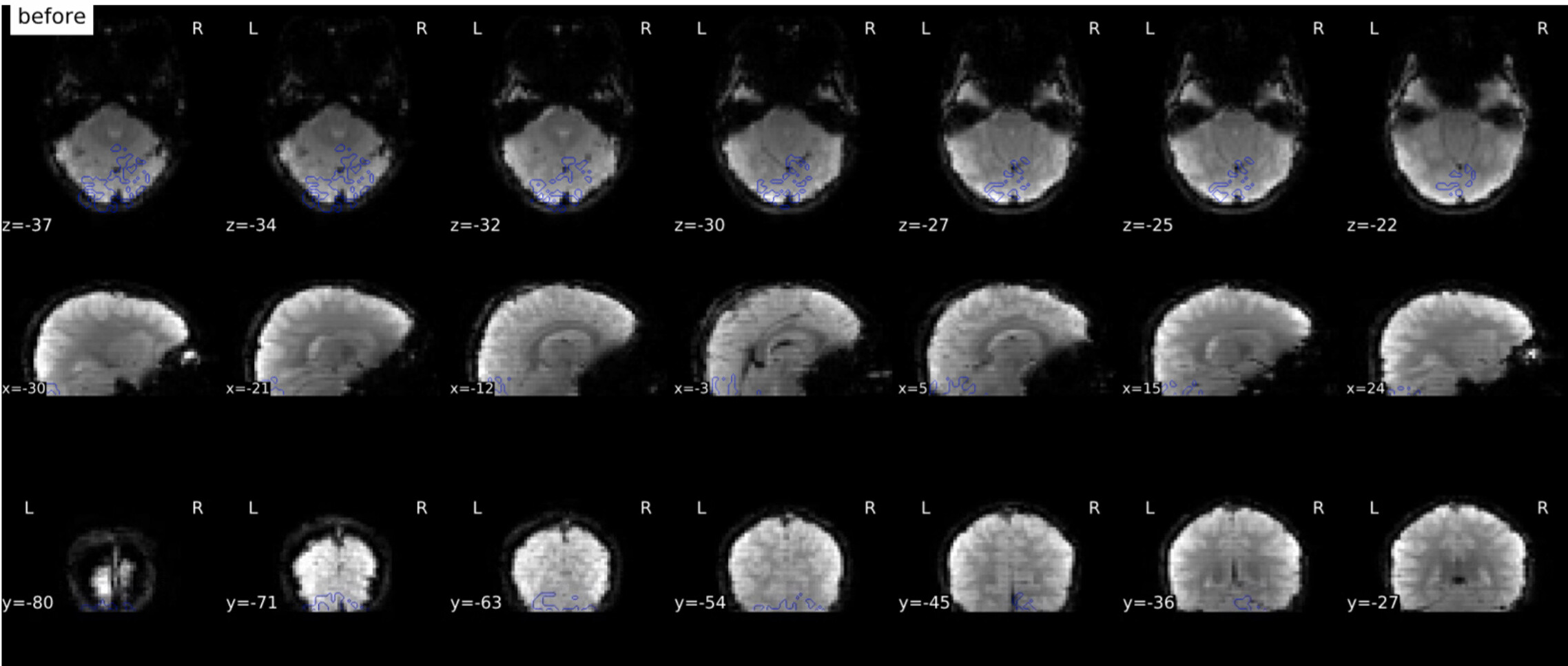
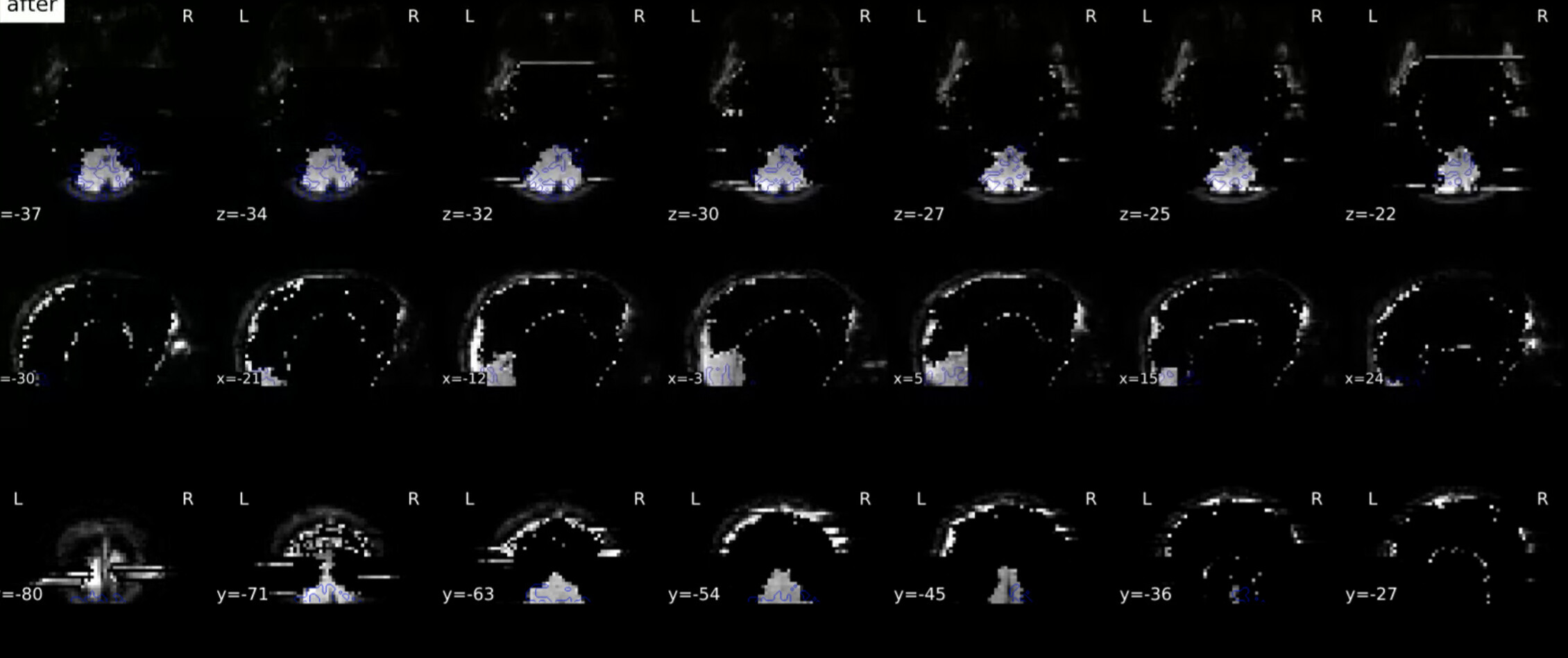
Thank you so much for looking into this. The SDC images I had are definitely not great, but they do look different from yours which also concerns me. Here’s a before and after.
Oh glad that it worked with that --ignore flag! I will definitely check my other subjects’ fieldmaps and metadata. Thank you so much figuring this out.
This very much looks like the fieldmap is unusable. I just learned yesterday that these are spiral echo fieldmaps. Therefore, the amount of preprocessing is very minimal - that doesn’t mean fMRIPrep did not fail, but there’s definitely smaller room for that than in other types of fieldmaps.
Would you suggest then keeping the fieldmaps for the subjects where SDC was successful and ignoring them for the subjects where the fieldmaps are unusable? Or, do you see potential problems in group-level analyses with a sample where some go through SDC and some don’t?
I guess that will depend on your analysis design. If you are planning on voxelwise analyses where misregistration errors between the anatomical and functional images are not critical, then you are probably fine. As a result, your outcome of interest will probably smooth out a little with this misregistration caused by SDs, especially on those regions most affected by distortions: vmPFC and temporal lobes. Please note that the whole brain will have misregistration, as the SDs are global although small, across the whole brain.
However, if you are planning on some surface-base analysis, I guess the misregistration between anatomical and functional is critical, so introducing artificial experimental conditions such corrected-vs-noncorrected will probably derail your experiment. This is because surfaces are very specific when sampling the functional signal, and in that case, the distortion introduced by B0 inhomogeneity may be comparable to the cortical thickness within which you are sampling.