I just found out about the rodent version of fmriprep! This is amazing! Is it already fully functional and what is the best way to run nirodents on a windows system? Can I use a virtual linux machine or is there a docker image available?
My name is Eilidh, and I am one of the team working on fmriprep-rodents and NiRodents.
These are technically two separate (but overlapping) projects; NiRodents is a toolbox for rodent image processing, which contains artsBrainExtraction, our brain masking tool. On the other hand, we intend for fmriprep-rodents to be similar to the original fMRIprep, in that it will automate the processing of rodent MRI images using many different tools, including those available in NiRodents.
To answer your questions: NiRodents is currently functional, and we encourage people to help us test it out! fmriprep-rodents is currently a work-in-progress, but we hope that it will be available for wider testing very, very soon.
To run NiRodents on a windows system, we recommend using Docker and pulling the latest image from nipreps/nirodents. The default entrypoint of the docker image is to run artsBrainExtraction by default, using a command like:
or alternatively, running the tool from the bash command within the image: docker run --entrypoint /bin/bash --rm -it -v /path/to/data:/data:ro -v /path/to/output:/out nipreps/nirodents:latest
When it ran the first time it gave me this message:
RuntimeError: An instance of template <tpl-Fischer344> with MR scheme 'T2w' could not be found
But it worked the second time! It took a while (15mins) to finish processing but ran through. However, in the end the images didn’t seem to fit the template quite as expected. I assume it’s because the template and input files do not have the same orientation. What orientation do I need for my input images and is there a specific tool you would recommend using together with artsBrainExtraction?
Great news that you got it to run, though I’m sorry the results weren’t as expected.
The error you posted is an issue we were aware of - it’s related to the downloading of the template from templateflow, and happens after the first time templateflow is called when the template needs to be downloaded. It should be fixed in future versions because we have updated the version of templateflow included in the docker image.
artsBrainExtraction expects images in RAS+ orientation, and I know there are some discrepancies with this respect in the rodent imaging community. The best solution is to use a Nifti conversion software which will correctly identify these orientations. I recommend BrkRaw for Bruker scanners, which also generates the file names to be BIDS-compliant (which is also necessary to take advantage of all the features of NiRodents).
Hi Eilidh,
I face some issues in running the bash command:
docker run --entrypoint /bin/bash --rm -it -v /path/to/data:/data:ro -v /path/to/output:/out nipreps/nirodents:latest
and the error is:
The command I gave above is intended to be a template. The most recent version of the docker image on dockerhub can be accessed by using the command docker pull nipreps/nirodents:0.2.1 which would then be run by using the command above. So nipreps/nirodents:latest and /path/to/data are just placeholders, which should be replaced with the respective identifier for the docker image (e.g. nipreps/nirodents:0.2.1) and the absolute path to your files stored locally.
As I said, I notice this version is a little out of date, so I’m grateful that you’ve brought this to my attention. I will try to finish some changes this week and upload a new version, but if you want to use the most up-to-date version in the meantime, I’d suggest you clone our github repo and build the docker image using the DockerFile there.
Hi Eilidh,
Thank you for your response, I could download the image finally and run the code below:
docker run --entrypoint /bin/bash --rm -it -v /path/to/data:/data:ro -v /path/to/output:/out nipreps/nirodents:0.2.1
and now I can run the command artsBrainExtraction, but I don’t know how i should address my file as input. as I am working in wsl2 I have a shared space between linux and windows in mnt folder. my files are in the /mnt/e/RAT_DATA/NSA/nsa01/anat/nsa01_anat.nii e.g., but when I enter this as input to the artsBraiExtraction I get following error:
traits.trait_errors.TraitError: The ‘reference_image’ trait of a _FixTraitApplyTransformsInputSpec instance must be a pathlike object or string representing an existing file, but a value of ‘/mnt/e/RAT_DATA/NSA/nsa01/anat/nsa01_anat.nii’ <class ‘str’> was specified.
May you help me with this error please?
And also thank you for mentioning the version of the tool. I am testing your tool for brain extraction of my rat data, if it works well on my data I will use the latest and most up-to-date version.
The issue is still with the Docker command, as artsBrainExtraction can’t find the images within the container. As I mentioned above, /path/to/data should be a placeholder for the directory where your files are stored.
The -v /path/to/data:/data:ro part of the command instructs docker to bind the volume (-v) /path/to/data to the container as /data, and it is read only (:ro) so as not to manipulate the original files. If /path/to/data doesn’t exist, there will be no files in /data. Consequently, there is no /mnt/e/RAT_DATA/NSA directory in the docker container, so, I’d suggest using the command:
docker run --entrypoint /bin/bash --rm -it -v /mnt/e/RAT_DATA/NSA/:/data:ro -v /mnt/e/RAT_DATA/NSA_derivatives/:/out nipreps/nirodents:0.2.1 and then run artsBrainExtraction -o /out/NSA_derivatives /data/NSA/nsa01/anat/nsa01_anat.nii
which writes outputs to /mnt/e/RAT_DATA/NSA_derivatives on your local machine.
I hope that this works well on your data, but there have been recent improvements which should help it run better if it does not. In particular, there have been changes to help when the mask includes some of the dorsal skull surface.
Hi @eilidhmacnicol . I’ve been following this thread to learn how to use nirodents. I am pretty excited about this tool.
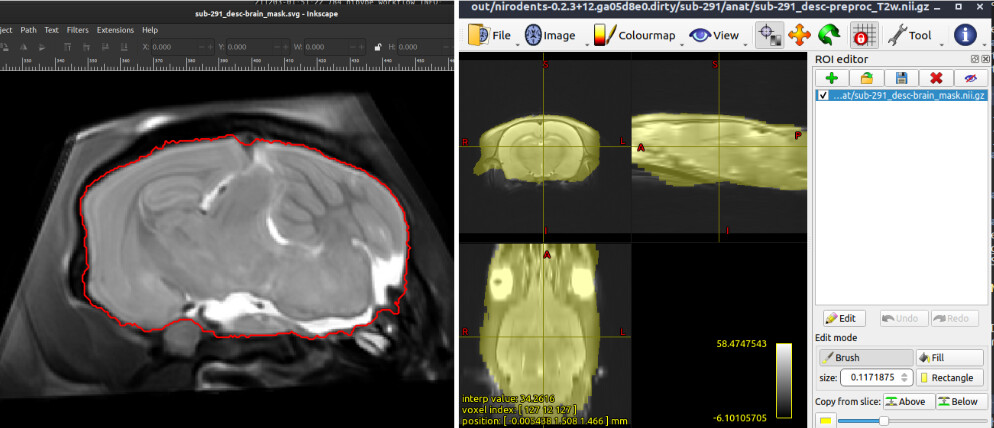
I am confused because the output mask seems to be an intermediary file, rather than the actual final mask. I assume this because the .svg files in /out/figures show a mask delimiting the brain (albeit in an oblique view, which I’ll figure out later), yet the file in /out/*brain_mask.nii.gz shows a mask that encompasses much more than the brain. See snapshots below, please.
Hi @lconcha and thanks for your interest in nirodents!
I have a couple of pointers:
We recommend viewing the .svg files in an internet browser because this will show a mosaic view in all three planes that alternates between the native space image and the template image warped to native space. If I was to guess, I would say that the image you can see is the warped template image, which explains the less-than-ideal result.
We only recommend using --antsAI in cases where images do not have the correct head orientation with respect to the template (e.g. some 2dseq to nii convertors output images that appear to have the AP and IS planes switched, which requires rotation of the image). As you used brkraw, I’m pretty confident that if you overlay the template image and the raw images, they will be in mostly the same orientation and this argument is not necessary. A further caveat: we did sink a lot of time in testing --antsAI settings, but we have not yet found the optimal settings for preclinical images (which is why we provide the options on the command line to tune the search settings).
Try running your results again without --antsAI, and view your results in your browser, and let me know how you get on. I appreciate your patience, as many of these issues can probably be explained in documentation which is a high priority for us in the near future.
Hello, I’m endeavoring to grasp the utilization of artsBrainExtraction within a Linux environment, but I’m encountering a recurring error message: The ‘reference_image’ trait of a _FixTraitApplyTransformsInputSpec instance must be a pathlike object or a string representing an existing file.