I’m a beginner in neuroimaging and have recently completed an ALE-based meta-analysis on self-referential processing. I now have unthresholded Z-value ALE maps, and I’m trying to decode them using Neurosynth with NiMARE’s continuous.CorrelationDecoder.
Could someone confirm if my approach is correct, and whether these unthresholded maps can be used directly with the Neurosynth dataset? Below is the code I am using. Any guidance would be greatly appreciated!
The code looks reasonable t ome, but if your goal is to decode ALE maps then I think it makes more sense to use ALE as the meta-analytic estimator within the CorrelationDecoder. I’ve never actually tried using a CorrelationDecoder with ALE, but it should work just fine.
I would recommend using the same settings for the ALE estimator that you used for your ALE meta-analysis as well.
from nimare.meta.cbma.ale import ALE
ale_estimator = ALE()
decoder = continuous.CorrelationDecoder(
feature_group=None,
features=None,
meta_estimator=ale_estimator,
target_image='z',
)
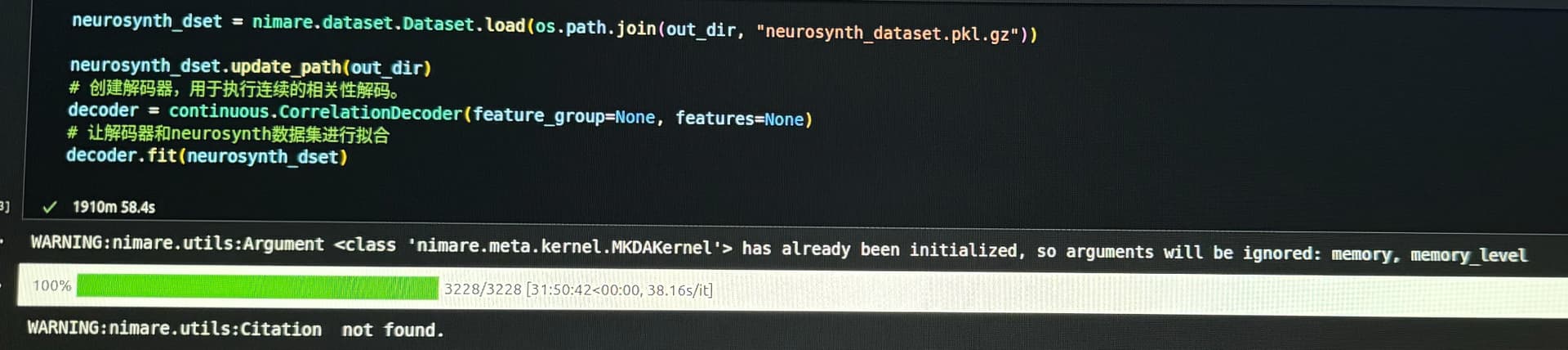
Thank you very much for your help. Before receiving your response, I had already run the code, but encountered the following error (screenshot attached). I’m not sure what caused it.
The first warning, about the MKDAKernel, can just be ignored.
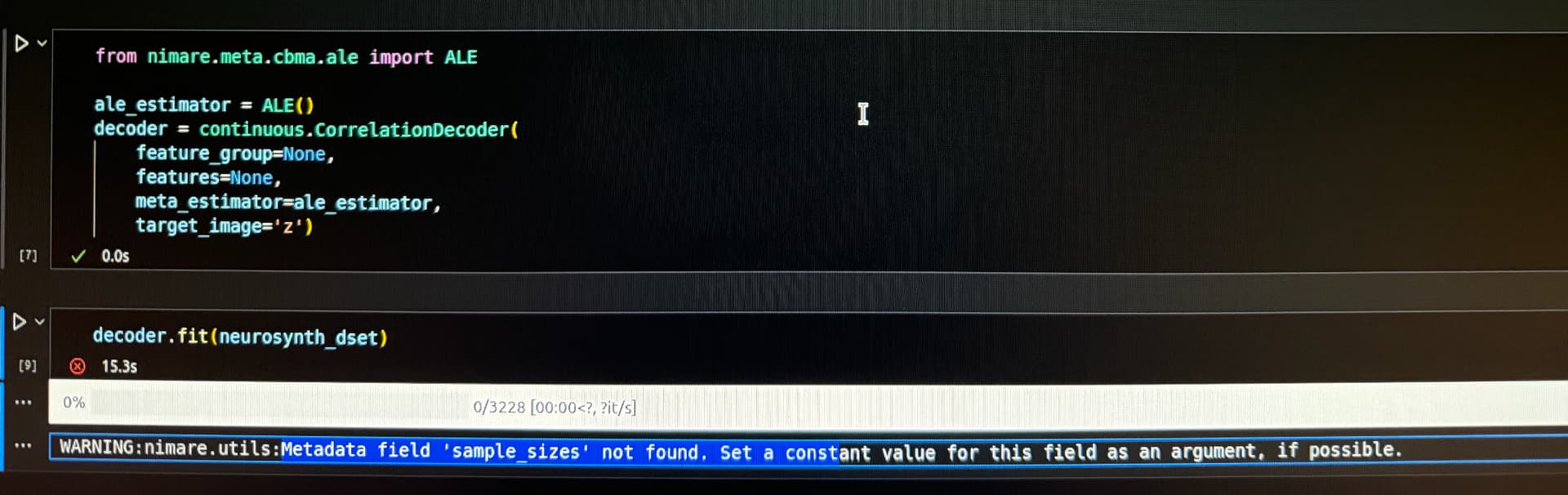
The ALE warning relates to the ALE algorithm, which determines the size of the Gaussian kernel for each study based on the number of subjects in that study’s sample. However, the Neurosynth database doesn’t have information about sample sizes, so you need to set a single sample size to apply to all of the studies. You can specify a single “sample size” to use for all of the studies in the Dataset by change ale_estimator = ALE() to ale_estimator = ALE(kernel__sample_size=30) (30 is probably a fine choice since most studies have small samples).
When I execute decoder.fit(neurosynth_dset) , I keep seeing warning messages: WARNING:nimare.utils:Metadata field 'sample_sizes' not found. I would like to know if these warnings can be safely ignored. Thanks again!