Hello everyone,
I have already get the whole-brain tractogram with QSIRecon (mrtrix_multishell_msmt_ACT-hsvs). It has generated the track file (*model-ifod2_streamlines.tck,10 million) and the corresponding sift2 weight for each streamline (*model-sift2_streamlineweights.csv). Then I want to use pyAFQ to select some bundle of interest (BOI) by custom waypoint ROIs and probmaps.
According to my understanding, when calculating the metrics of a fiber bundle, the weighted average based on sift2 weight is required. So, the question is how can I calculate the scalar metrics (e.g. FA, MD) for each streamline and each bundle of my BOI with the streamline weight from sift2? Or, could it be possible to combine the sift2 weight and streamline together during the pyAFQ process of filtering the fiber bundles, and perform the filtering simultaneously?
That is a great question. I am not sure what a weighted average based on sift2 is “required”, but on the face of it, using sift2 weights seems like a great idea, and I would be quite interested to learn how/whether tract profiles that are weighted by sift2 weights differ from those that are weighted by the deviation of the streamline from the core of the tract (which is pyAFQ’s current default).
Fortunately, this should be possible to calculate with currently available software: The DIPY afq_profile function (see example here) takes an optional 'weights' key-word argument. I believe that if you provide the sift2 weights as an array that is ordered in the same order as your streamlines, the result would be a tract profile that is weighted proportionally by the sift2 weight assigned to each streamline.
I’d be quite interested to see a comparison if you conduct one.
Thank you very much for your kind reply. I agree that using the DIPY afq_profile function for weighted calculation of streamlines seems to make sense. But before that, we need to determine the index of the BOI streamlines in the original whole-brain tractogram, so that we can extract the sift2 weight in the right order. This is the problem that troubles me .
To achieve this, I have tried to write the original index into the metadata (data_per_streamline) of the whole-brain tractogram (both in .tck and .trk format) with DIPY. And then the tract file was provided to pyAFQ for bundle recognition, with the expectation that it can be filtered while carrying the index.
# Codes for writing the original index into the metadata
import numpy as np
from dipy.io.streamline import load_tractogram, save_tractogram
raw_file = "*desc-tracks_ifod2_100k.tck" # The same procedure applies to.trk files as well.
out_file = "*desc-tracks_ifod2_100k_withidx.tck"
sft = load_tractogramn(raw_file, reference="*space-T1w_FA.nii.gz"
n_streamlines = len(sft.streamlines)
orig_idx=np.arange(n_streamlines)
sft.data_per_streamline["orig_idx"] = orig_idx
save_tractogram(sft, out_file)
Unfortunately, it failed. I found that the .tck format does not seem to support metadata writing (this information is automatically erased during saving with DIPY save_tractogram function). Although the .trk format support metadata writing, it cannot be retained after pyAFQ bundle recognition…
Now I have another idea: For each streamline in the BOI, compare it one by one with the streamlines in the original whole-brain tractogram, thereby determining its index. But this seems to involve a relatively large amount of calculation (thousands of people * 10 million streamline). Do you think this plan is feasible? Or are there any other better alternatives?
By the way, although I had not thought about making a direct comparison between tract profiles that are weighted by sift2 weights and those under pyAFQ method before (I had only highly appreciated the bundle cleaning process of pyAFQ before), I now realize it is interesting. So after finding out the solution of the above problem, I’d like to do the comparison and share with you all
The recognize function, which is the work-horse of pyAFQ bundle recognition, has a return_idx key-word argument that is designed specifically for this kind of use-case, where indices in the original tractogram need to be propagated. This does mean that you will have to set up everything and call it directly rather than depending on pyAFQ’s data management and workflow API. Does that provide a good enough solution?
Correction: I think that I might be wrong, and you can actually pass return_idx=True to the GroupAFQ class and also set that up as part of the config file, if you are using the CLI. I believe that this would save the original indices in the individual tract trk files, just like what you were trying to do. So no need to write all the low-level code
Yes, just as you noticed, I also found that the clean function also has a return_idx parameter.
return_idx: bool
Whether to return indices in the original streamlines. Default: False.
In general, the cleaning process needs to be completed through multiple iterations (defaults to 5 iterations in pyAFQ). So the indices recorded in the log file seem to be relative to the most recent tractogram iteration, as shown in the following screenshots.
It seems that some modifications to the code of pyAFQ are necessary (although for me, a novice in coding, this might take some time). Nevertheless, this is indeed a promising discovery.
Hello Wei,
What version of pyAFQ are you running? I believe this is an error from an older version of pyAFQ as we no longer print “Removed indices” we print “Kept indicies”. I tested this on the latest pyAFQ version and it seems to work. Try updating to a newer version and see if this helps. Let me know how it goes.
However, these kept indicies in logfile (DEBUG:AFQ) do not seem to be the ones in the original whole-brain tractogram that I estimated using another pipeline and then fed into pyAFQ workflow for bundle recognition. Then I try to specify the return_idx argument in the workflow (ParticipantAFQ) like this:
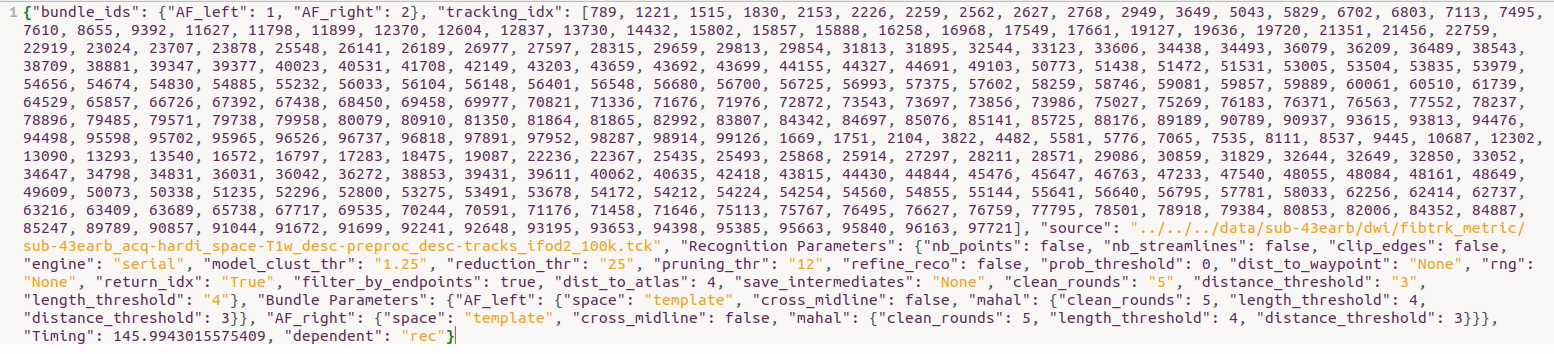
Is my parameter configuration correct? It completed smoothly and I found the “tracking_idx” field in the result file “sub-*_acq-hardi_coordsys-RASMM_trkmethod-probCSD_recogmethod-AFQ_tractography.json” as below.
Are these indices relative to the indices in the original tractogram? If so, is there a parameter setting that can output the indices for different bundles separately? And by the way, what are the definitions and relationships between these three indices (i.e. 1-"Kept indicies" in logfile (DEBUG:AFQ), 2-“tracking_idx” field in the result .josn file, 3-indices in the original tractogram)?
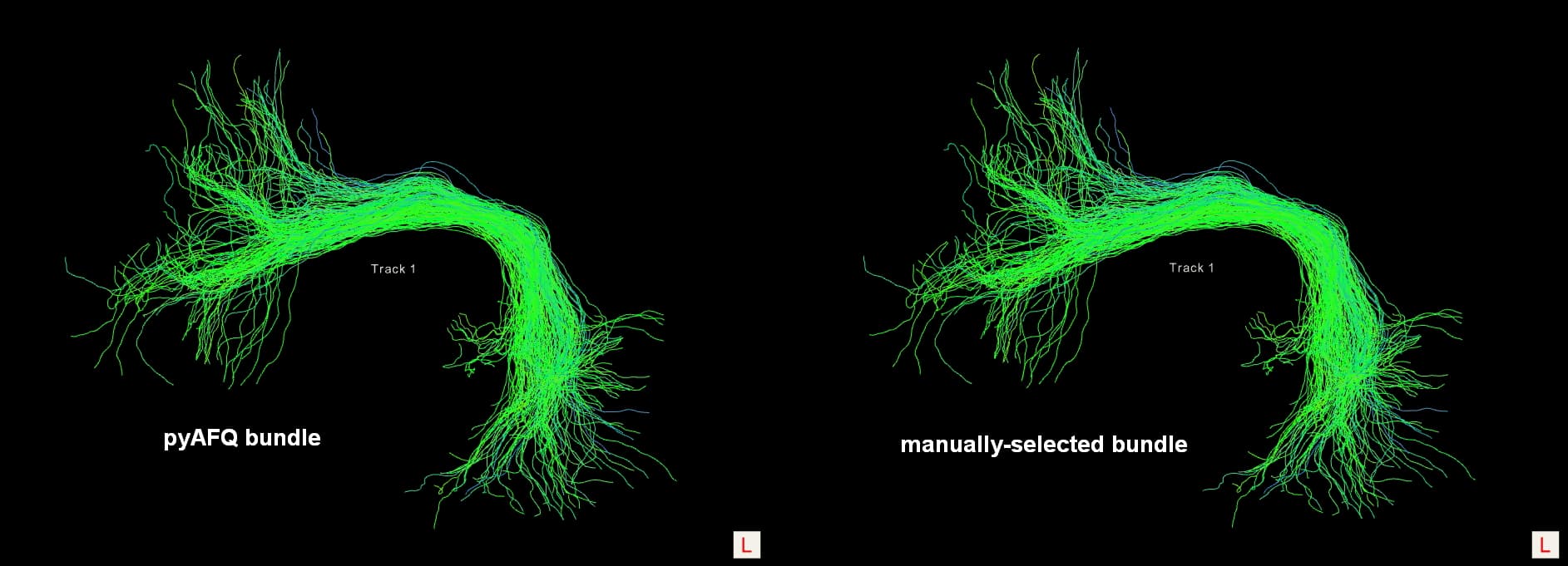
In order to verify the correctness of the indices (i.e. the “tracking_idx” field in the result file “sub-*_acq-hardi_coordsys-RASMM_trkmethod-probCSD_recogmethod-AFQ_tractography.json”) generated with the return_idx argument, I manually selected the streamline set from the original whole-brain tractogram according to those indices using DIPY library. Then I compared the coordinates of streamlines between the streamline set I selected and the bundle recognized by pyAFQ. Unfortunately, nearly one-third to one-half of the streamlines are not matching! However, visually, the two bundles appear to be similar.
I’m confused about where the problem lies. Is there a bug in the indices generated by pyAFQ, or did I make a mistake in my inspection process?
I should have said earlier: these DEBUG indices from the logfile are really only useful for debugging. They are indices relative to what the cleaning received, not the original tractogram. So ignore (1). However the tracking_idx you found in the JSON file should be indices into the original tractogram. So (2) and (3) should be the same. So, manually selecting from the original tractogram using tracking_idx should have worked. So I am not sure what is wrong.
One question: do they visually look similar or the same? Because one thing pyAFQ does is flip streamlines so they are oriented in the same direction. So roughly half of the streamlines in the bundle output by pyAFQ should be flipped relative to the original tractogram. If you account for the flipping, the streamlines should match exactly.
Correction for previously inaccurate statement: the two bundles visually look the same. And thanks for pointing out the “streamline flipping” procedure in pyAFQ. The streamlines now matched exactly under a floating-point error tolerance of 1e-4 when accounting for the flipping.
Some additional details hope to confirm with you:
(1) Considering fiber bundle format conversion, is a tolerance of 1e-4 acceptable? (pyAFQ bring .tck to .trk; the manually-selected bundle saved in .tck)
(2) Does “streamline flipping” procedure affect subsequent streamline/bundle-level analysis (e.g., calculating streamline/bundle metrics based on SIFT2 weights, or extracting metrics for specific segments of streamlines/bundles)? Or does the subsequent analysis require additional consideration of the “streamline flipping” procedure?
(3) When identifying multiple fiber bundles simultaneously, is there a parameter setting that can extract the streamline indices corresponding to each individual fiber bundle separately, rather than storing them all in “tracking_idx” (as shown below)? If not, what is the best way to manually separate them?
(1) I am not entirely sure where these differences come from, but I think that 1e-4 differences are acceptable. For consideration of this: assume that the coordinates are in units of voxels, and assume a voxel of 2 mm isotropic. This means that a 1e-4 difference is a difference of 0.2 microns between the coordinates in the two files. I doubt that the measurement has the precision where 0.2 microns makes any practical difference.
(2) I am not sure that I understand the question. Let me say this: The flipping is specifically intended for the purpose of extracting tract profiles from the tracts and maps of tissue properties such as FA/MD/etc. The reason we do this is that we collect information from the maps along the length of each streamline and we want them all to be co-extensive in space when we do that, so that we don’t start collecting from one end of the tract in one streamline and from the other end of the tract for another streamline. Not sure, did this answer your question.
(3) Not with current versions, but we’re working on it for future versions here. For now, you will have to rely on the order of the tracts in “bundle_ids” and the number of streamlines in each tract to know where one tract starts and ends in “tracking_idx”.
Thank you and John. I think these did answer my questions.
Additional explanation for (2): I am considering performing analyses beyond tract profiles, such as calculating overall FA/MD of bundles recognized by pyAFQ combined with MRtrix SIFT2 weights. As you mentioned, flipping merely alters the spatial orientation of streamlines to make them spatially consistent, so I agree this procedure should not impact other analyses.