Hi everyone,
We are working on fetal brain MRI acquisition and data analysis recently. And we tried to use the dHCP-structural-pipeline to analyze the fetal brain MRI data.
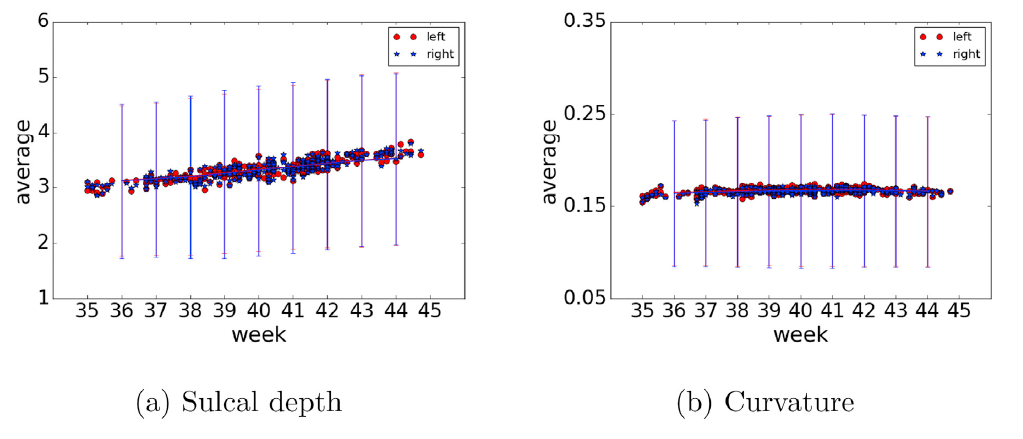
However, we found that the results of sulcal depth and curvature are quite strange. With our data, the average sulcal depth is 19~31 (unsure what the unit is), which is almost 10 times larger then the actual depth. The average curvature ranges from 1.9 to 2.6, which is 10 times larger than the values reported in some papers. We’ve carefully checked the segmentation accuracy and it looks reasonable.
In addition, to make sure that there is nothing wrong with our data, we tried 38w dhcp data(rel-2), and got a sulcal depth of 26.83629 and the curvature of 2.453721.
Dose anyone meet the same situation? Is there any further calculation requested? Any advice may help, thanks in advance!
I used the workbench viewer to check my results, however the results seem pretty reasonable. So I think the problem maybe in dHCP-structural-pipeline-measures.
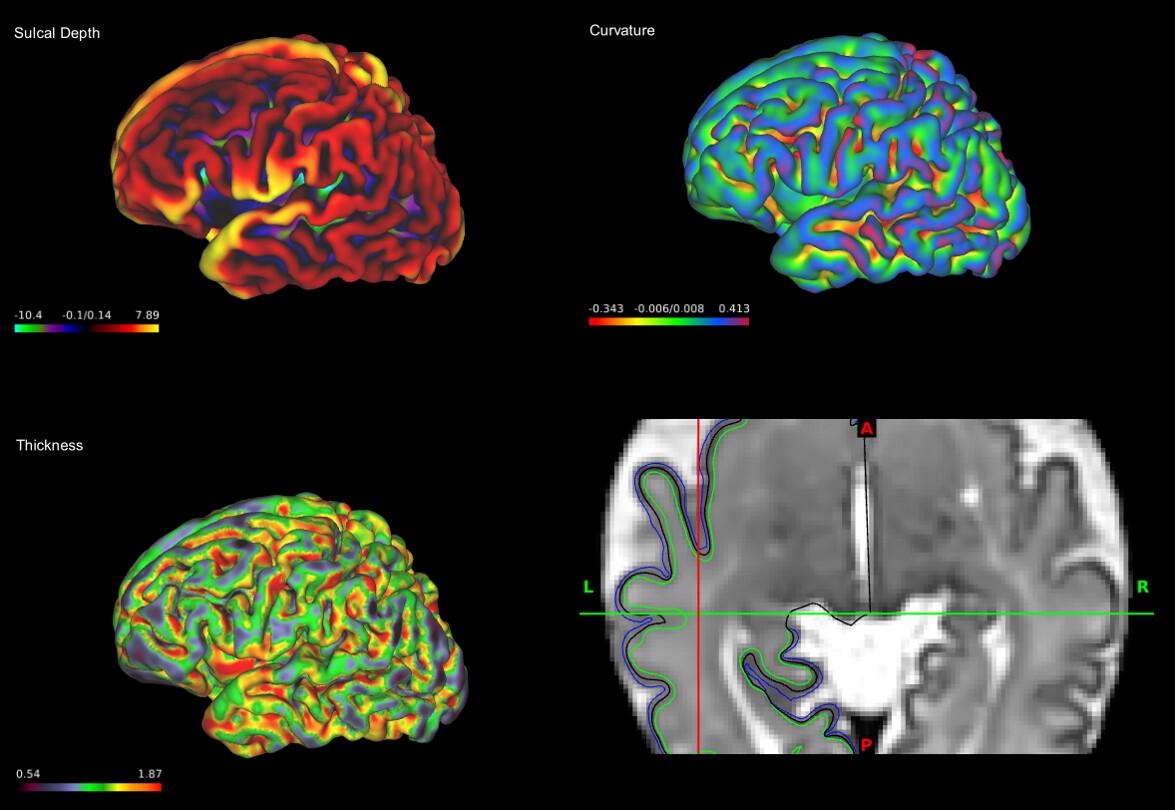
I have carefully checked a few subjects from the second dhcp release (screenshots attached). Both @lzjwilliams and I think that the values of the curvature and sulcal depth measures looked reasonable and consistent. I also found that the range values of the sulcal depth are similar to this paper:
It is worth checking the histogram for each metric and see the whole distribution and not only the range of values in case there are any outliers. It is also important to check the inflated surfaces, as some of the metrics are calculated from the inflated (deformed) surfaces.
For example, here are how the curvature and sulcal depth measurements are calculated in the dhcp pipeline:
Surface curvature is estimated from the mean curvature (or average of the principal curvatures) of the white matter surface
Sulcal depth is estimated during inflation using maps of mean convexity/concavity. This represents the change of a node’s position in normal direction, and is proportional to the depth of major sulci
Thank you for your detailed reply!
I have read those two dhcp papers again, and here are some questions need discussing:
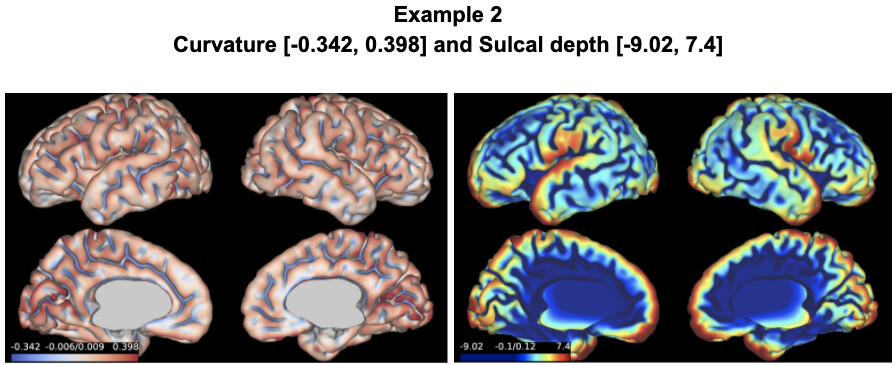
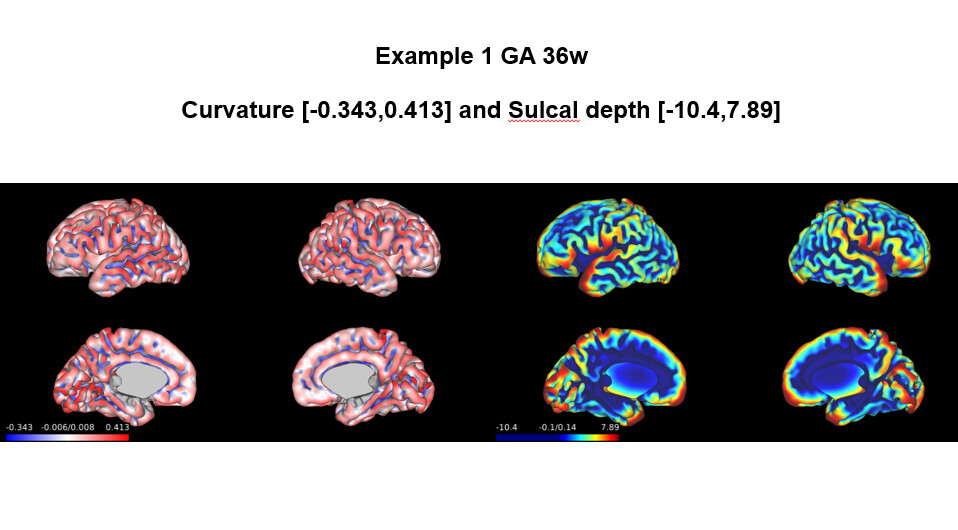
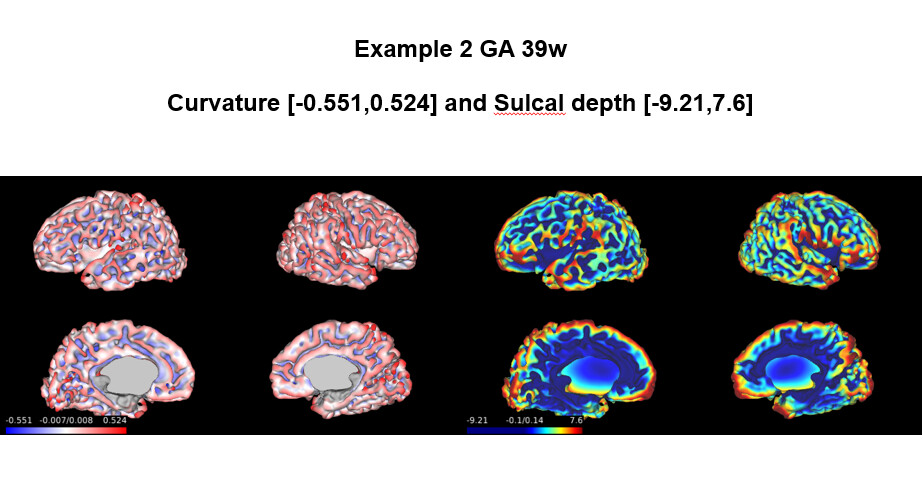
The results of my fetal data showed in Example 1 and 2 are reasonable, just like the examples you showed. However, in the …/reports/pipeline_all_measures.csv , the results of sulcal depth and curvature are as follows:
Yes, I run the biomedia/dhcp-structural-pipeline-measures:latest in docker by myself. And I used the results calculated by biomedia/dhcp-structural-pipeline:latest in docker to run the measurement pipeline.
Will refining the cGM label in tissue_labels.nii.gz be helpful for getting more accurate results? Or is there another better way to refine my results?
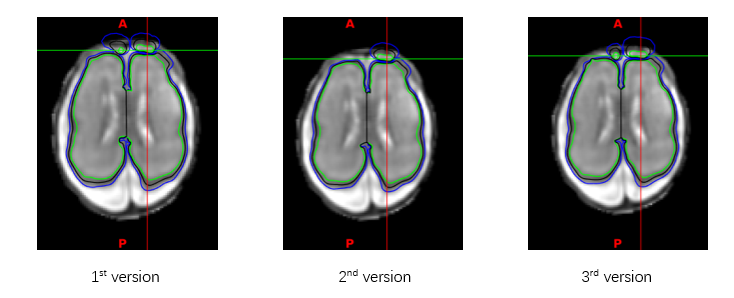
I tried three different version of tissue_labels.nii.gz, 1st version without manual refinement after segmentation, 2nd version with manual refinement, 3rd changed the specific slice. But the results of dhcp-structural-pipeline seem unaffected.
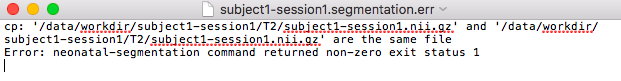
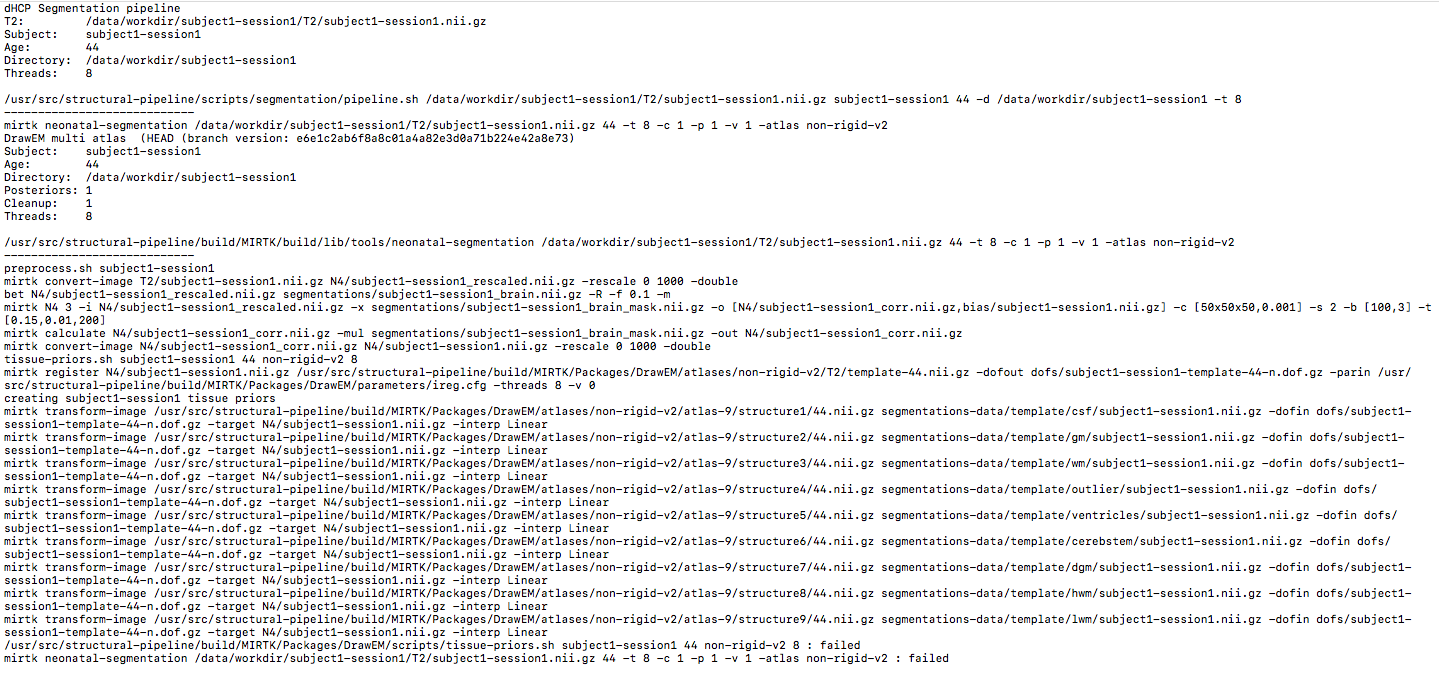
I suppose you are succeed to run pipeline from docker hub. So, I have a question, when I run pipeline, I have an issue:
Pipeline failed: see log files/data/logs/subject1-session1.segmentation.log /data/logs/subject1-session1.segmentation.err for details
Would you be able to fix it please?
Currently there is no easy way to run the pipeline with manually corrected labels although we have been talking about it as a way to fix some problems we are having with clinical datasets. I’m afraid I cannot give you a timeline for that however.
Could you please attach the /data/logs/subject1-session1.segmentation.err and /data/logs/subject1-session1.segmentation.log? So I can see which step went wrong.
Thank you for your reply.
I did segmentation with my data by using DrawEM independently, and put the manually corrected segmentation results in the right folder path required by dhcp-structural-pipeline. And then I run the dhcp-structural-pipeline in docker. I suppose the pipeline will skip the segmentation step and do the rest of the pipeline, like reconstruction of surfaces with the right labels.
It’s been a while since your last reply. Is there any new update about the measurement pipeline? Are those sulc and curvature results reliable?
Look forward to your reply. Thank you in advance!
Sorry for the late reply.
I take a look of my successful data, it seems that the last step in /usr/src/structural-pipeline/build/MIRTK/Packages/DrawEM/scripts/tissue-priors.sh failed. It should be like this: mirtk calculate segmentations-data/tissue-posteriors/gm/subject1-session1.nii.gz -mul 100 -out segmentations-data/gm-posteriors/subject1-session1.nii.gz.
I have never met this problem with my data, so I am not sure what cause this.
But maybe you can try some other data to see if it was the data’s problem.
Do you mean those gifti files or the spec file in ..\derivatives\sub-subject1\ses-session1\anat\Native ?
And are there any recommended toolboxes I can use to get those measurements in the files, like the ones in pipeline_all_measures.csv?
Thanks a lot!
I am working to analyze the midthickness surface curvature of the dHCP data release 2. However, I am not able to find the cortical surface files (surf.gii) in the file tree of the data provided via GIN. Is there any way to obtain the surface files for data release 2?
The GIN repo is specific to the SLCN challenge that @EmmaR and I are running. If you want the midthickness files, and other cortical data, you will need to download it from the dHCP website.